SINDROME DI APERT
La Sindrome di Apert venne descritta per la prima volta nel 1906 dal neurologo francese E. Apert. Questa malattia è la conseguenza della mutazione in un singolo gene localizzato sul cromosoma 10q. Ad oggi ancora non è noto come tale mutazione avvenga. La trasmissione della malattia avviene in modalità autosomica dominante ed ha un incidenza tra 1:60.000 e 1:120.000 nati.
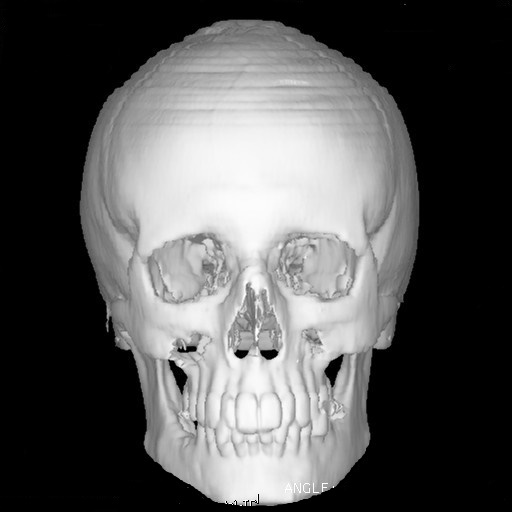
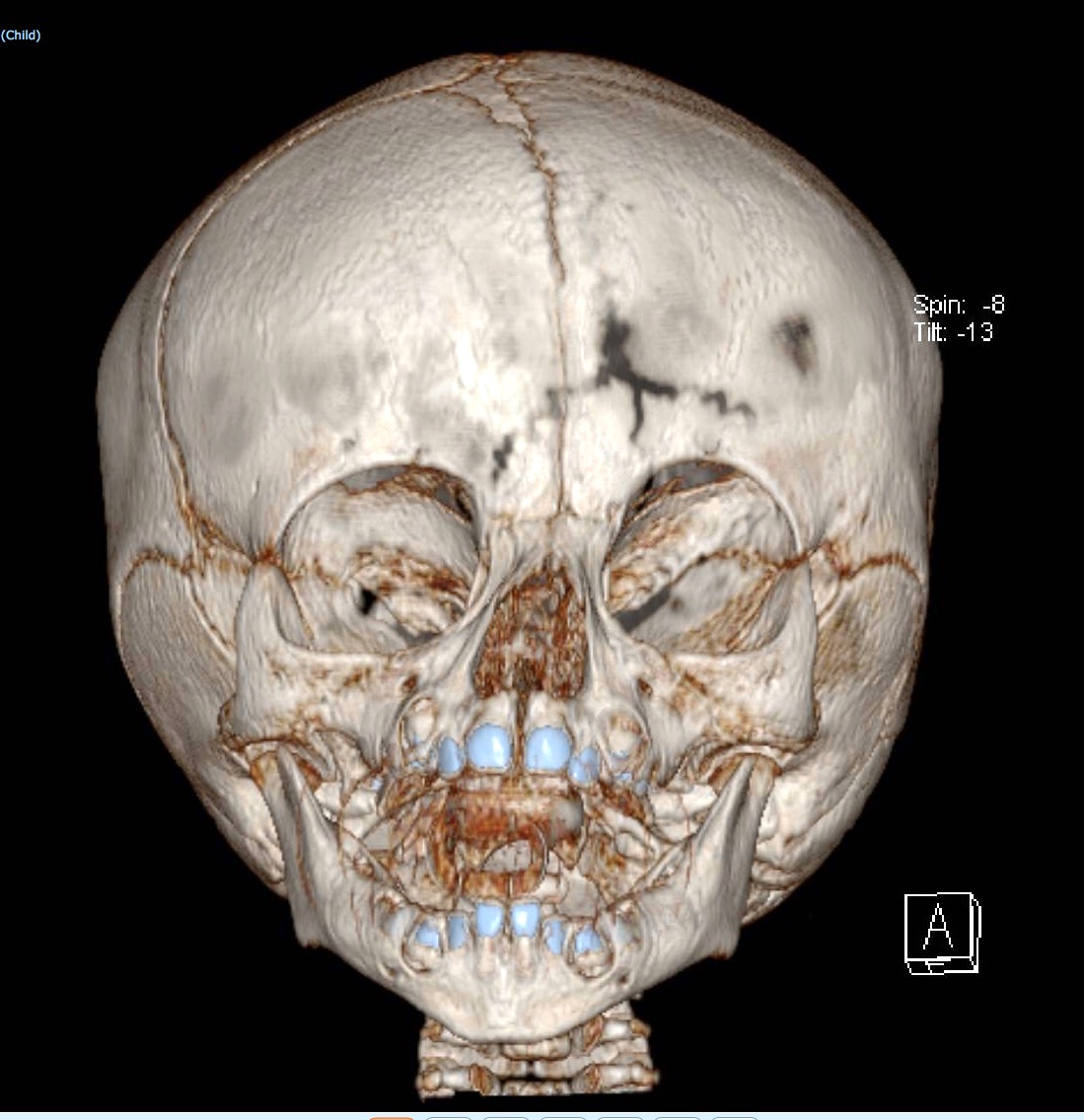
Nella sindrome di Apert anche tutte le suture del cranio possono essere prematuramente chiuse. La deformazione del cranio che ne deriva è caratteristica: fronte molto alta (turricefalia), mascellare arretrato con inversione dell’occlusione tra le arcate alveolari superiore ed inferiore, orbite corte con occhi sporgenti (proptosi o esoftalmo) e ad una distanza tra loro superiore alla norma (ipertelorismo), regione temporale convessa. A volte la deformità del cranio è molto grave ed assume la cosiddetta forma a “trifoglio”. L’ipertensione endocranica è sempre presente e molto frequente è la comparsa di idrocefalo.
Altra anomalia sempre presente nella Sindrome di Apert è la sindattilia di mani e piedi che coinvolge tutte le dita conferendo sia alle mani che ai piedi una forma caratteristica.
Il trattamento chirurgico della Sindrome di Apert prevede l’esecuzione di numerosi interventi chirurgici, alcuni in equipe con i neurochirurghi. La prima operazione, da eseguire tra il 4° ed il 6° mese di vita, consiste nell’avanzamento fronto-orbitario allo scopo di ridurre la pressione intracranica e dare più spazio agli occhi all’interno delle cavità orbitarie. Il secondo intervento si effettua tra il 5° ed il 6° anno di vita e consiste nell’avanzamento del mascellare, delle orbite e della fronte. Anche questa operazione viene eseguita in equipe con i neurochirurghi. Il trattamento chirurgico successivo varia in rapporto alla crescita della faccia e della mandibola.
La correzione della sindattilia viene iniziata precocemente, già durante il primo anno di vita, in particolare se il primo dito della mano (il pollice) è coinvolto.